
Question: How Should Sponsors, Investigators, and IRBs Navigate the FDA’s Updated IND Safety Reporting Guidance?
Answer:
Accurate and timely reporting of safety events in clinical trials is essential for protecting participants and maintaining compliance. Sponsors, investigators, and IRBs each play a vital role in this process. To provide clarity, the Food and Drug Administration (FDA) issued two final guidance documents in December 2025, detailing safety reporting responsibilities for sponsors and investigators.
“Investigator Responsibilities — Safety Reporting for Investigational Drugs and Devices,” clarifies how [21 CFR 312.32], [21 CFR 312.64], [21 CFR 312.66], and [21 CFR 56.108] intersect in determining reporting responsibilities in drug trials.
- Under [21 CFR 312.32(c)], sponsors must submit an Investigational New Drug (IND) Safety Report to the FDA and all participating investigators within 15 days when the sponsor determines that an event meets the criteria in [312.32(c)(1)(i–iv)].
- Under [21 CFR 312.64(b)], investigators must immediately report all “serious adverse events” to the sponsor — whether drug related or not — and must assess the reasonable possibility of causality.
- Under [21 CFR 312.66], investigators must promptly report to the IRB all “unanticipated problems involving risks to subjects or others.”
- Under [21 CFR 56.108], an IRB must have written procedures that ensure prompt reporting to the IRB of any “unanticipated problems involving risks to subjects or others.” The IRB must also have procedures for reporting these unanticipated problems to institutional officials and the FDA.
Relationship between [21 CFR 312.64(b)] and [21 CFR 56.108]
[21 CFR 312.64(b)] requires investigators to report any “serious adverse events” to the sponsor, whether drug-related or not. The majority of these events will not meet the criteria outlined in 21 CFR 56.108 for reporting “unanticipated problems involving risks to subjects or others” to the IRB, as most are either anticipated or unlikely to be reasonably related to the drug.
| Who must report | What | To whom |
|---|---|---|
| Sponsor | IND safety report [312.32(c)(1)(i–iv)] | FDA, Investigators |
| Investigator | Serious adverse events | Sponsor |
| Investigator | Unanticipated problems involving risks to subjects or others | IRB |
| IRB | Unanticipated problems involving risks to subjects or others | Institutional officials and the FDA |
Relationship between [21 CFR 312.32(c)], [21 CFR 312.66], and [21 CFR 56.108]
[21 CFR 312.32(c)] specifies evidence criteria for IND Safety Reports:
- A single occurrence of an event that is uncommon and known to be strongly associated with drug exposure [312.32(c)(i)(A)].
- One or more occurrences of an event that is not commonly associated with drug exposure but is otherwise uncommon in the population exposed to the drug [312.32(c)(i)(B)].
- An aggregate analysis of specific events observed in a clinical trial that indicates those events occur more frequently in the drug treatment group [312.32(c)(i)(C)].
- Findings from epidemiological studies, pooled analysis of multiple studies, or clinical studies that suggest a significant risk in humans exposed to the drug. Ordinarily, such a finding would result in a safety-related change in the protocol, informed consent, or investigator brochure [312.32(c)(ii)].
- Findings from animal or in vitro testing that suggest a significant risk in humans exposed to the drug. Ordinarily, any such findings would result in a safety-related change in the protocol, informed consent, or investigator brochure. [312.32(c)(iii)]
- Any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. [312.32(c)(i)]
The FDA has stated in the new guidance that the events that are captured in IND Safety Reports under these regulations meet the requirement in 21 CFR 56.108 for reporting “unanticipated problems involving risks to subjects or others to the IRB.”
Simplified Guidance for Sponsors: IND Safety Reporting and Decision Flowchart
The FDA’s updated guidance, “Sponsor Responsibilities —Safety Reporting Requirements and Safety Assessment for IND and Bioavailability/Bioequivalence Studies,” provides critical clarity on safety reporting obligations for sponsors. This guidance outlines the criteria for submitting IND safety reports and emphasizes the importance of thorough safety assessments in clinical trials.
To make these requirements more accessible, we’ve included a simplified version of the FDA’s decision flowchart below. This visual tool is designed to help sponsors quickly determine when and how to report safety events, ensuring compliance with regulatory expectations.
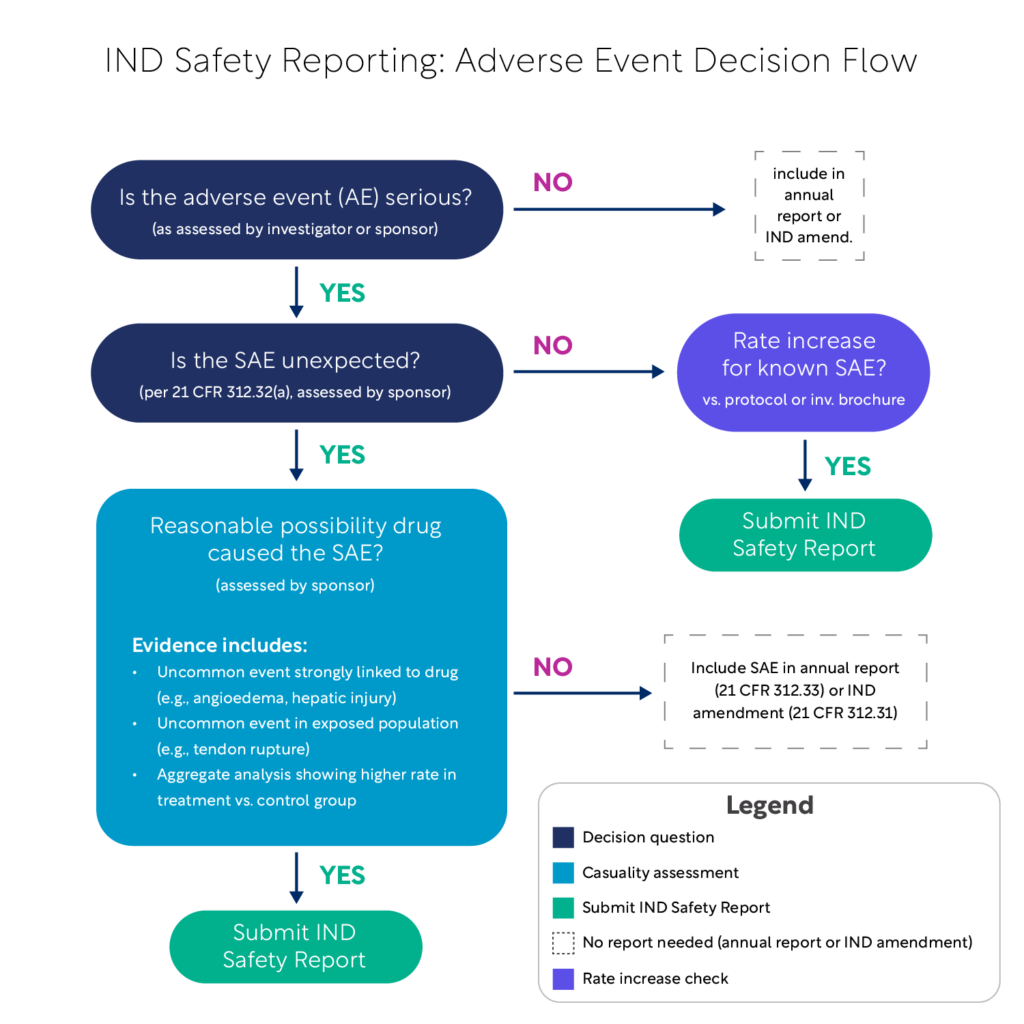
Streamlined Safety Reporting: Updated Resources for Compliance
The FDA emphasizes that investigators “should be familiar with and adhere to the IRB’s written procedures for reporting these unanticipated problems to the IRB.”
To support compliance with these requirements, WCG has updated its safety reporting guidelines to streamline compliance and support your team in managing these critical updates effectively.
| Document | Date | Requirement |
|---|---|---|
| WCG Guide for Researchers | 4/1/2026 | Safety reporting guidance can be found on page 62. |
| IRB.POL.HRP.071 | Updated 4/26/2026; revised 5/6/2026 | IND Safety Reports should be submitted to the IRB within five calendar days. Adverse events or SUSARs that require a protocol or consent change should be submitted to the IRB within five calendar days. |
| PRI Submission Forms | Updated in portals | List the following as promptly reportable: IND Safety Reports; any AE or SUSAR that requires a protocol or consent change. |
FAQs
Question:
Section VI of the investigator guidance states, “The investigator must submit IND safety reports to the IRB (§ 312.66) because FDA considers safety information that meets the IND safety reporting criteria under § 312.32(c) to be an unanticipated problem involving risk to human participants or others.” Up until this time, we [the CRO] submitted IND safety reports to WCG on the sponsor’s behalf only if there was a change in the conduct of the study (protocol, IB, ICF). Please clarify if the new guidance changes this rule and if all SUSARs from clinical trials should be submitted to WCG.
Answer:
No, the revised guidance does not require that all Suspected Unexpected Serious Adverse Reactions (SUSARs) from clinical trials be submitted to the IRB. However, the revised guidance does require that all IND Safety Reports be submitted to the IRB because the FDA has stated that they satisfy the criteria for an unanticipated problem.
Question:
In multicenter studies where sponsors submit safety reports on behalf of investigators, should they report only external adverse events and SUSARs that qualify as unanticipated problems, or all IND safety reports (SUSARs)?
Answer:
Sponsors submitting on behalf of investigators should submit all IND safety reports to WCG’s IRB. Item 11 of the promptly reportable information list in the WCG Guide for Researchers (Rev 1.29) now explicitly includes “All IND safety reports,” reflecting the FDA’s position that every IND safety report meeting the criteria under 21 CFR 312.32(c) represents an unanticipated problem requiring IRB reporting.
The IRB also requires reporting of any adverse events or SUSARs that require a change to the protocol or consent. WCG’s position is that it is the sponsor’s responsibility to determine if a SUSAR is an IND safety report.
Further, footnote 18 of the FDA guidance states:
Many study protocols specify that the sponsor will submit IND safety reports to the IRB on the investigator’s behalf. In these situations, where the investigator receives confirmation that the sponsor has sent the report to the IRB, FDA would not expect an investigator to provide the IRB with a duplicate copy of the report. Such an agreement should be documented.
Question:
Does WCG’s policy for reporting safety events apply exclusively to investigators reporting events occurring at their own sites?
Answer:
The five-calendar-day reporting window for promptly reportable information outlined in the WCG Guide for Researchers applies to all submissions – including IND safety reports – regardless of whether the event occurred at the reporting investigator’s site or at another site in a multicenter study. When a sponsor submits on behalf of investigators, the same five-calendar-day timeline applies to those submissions, calculated from the date the submitter becomes aware of the event.
Question:
We’ve recently noticed notifications from WCG IRB regarding submitted SUSARs, indicating whether the board is reporting them to the FDA. These notifications seem to reflect a change in process, as we had not received similar updates in the past. Could you clarify the review process for these notifications, including any associated timelines or criteria for determining when reports are submitted to the FDA?
Answer:
Process Change: WCG updated our policies in April 2026 in response to the December 2025 FDA guidance.
Timelines: A subject matter expert generally reviews IND Safety reports within one week of receipt. Board reviews of this type of issue generally occur on Wednesdays. Letters are sent within a few business days of the meeting.
Reporting: WCG will default to reporting to the FDA unless we have documented evidence that the FDA has already been notified. In cases where we have that evidence, we will not continue to report the issue to the FDA.
Question:
Does the Board review information in an IND safety report to determine if it indicates an increased risk in the study? If so, does this review include assessing whether actions are needed by the investigator to protect the rights and welfare of participants?
Answer:
The FDA has determined that IND safety reports are unanticipated problems. WCG’s IRB reviews the information submitted and considers what, if any, additional actions are required to maintain approval. The actions required, or that no additional action is required, are included in the Board letter.
Question:
If we [the site] send in an IND safety report that the sponsor has already submitted, is it considered an unanticipated problem involving risk to human participants or others?
Answer:
WCG’s IRB reviews the information and, if found to be duplicative, will note it and not send it for Board review.
WCG IRB knows what it takes to navigate evolving requirements with confidence. And knowing changes everything.
Don't trust your study to just anyone.
And we’re the best for a reason. Experience the WCG difference starting with a free ethical review consultation. We’re here to help you streamline, alleviate, and accelerate.