
On September 30, 2021 the FDA published a new draft guidance, Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial[1]. This draft guidance describes recommended approaches for sponsors conducting early stage umbrella trials of gene therapy products for a single indication in a defined population; the draft guidance focuses on relationships among multiple Investigational New Drug (IND) applications applied to a single master clinical trial protocol. The recommended scheme should be very useful for sponsors planning new submissions and amendments for this type of trial.
Per an existing FDA definition, an umbrella trial is “designed to evaluate multiple investigational drugs administered as single drugs or as drug combinations in a single disease population.”[2] The present draft guidance is specifically intended for early stage umbrella trials where multiple versions of a cellular or gene therapy product are being studied in a single disease population, and the products are generally submitted in separate INDs that are cross-referenced among each other. Note that some umbrella trial designs, especially those involving late-stage products, may include a sorting step where participants are segregated into different study arms based on one or more biomarkers[3]; such segregation does not apply to the present draft guidance.
The draft guidance includes useful notes and an appendix with examples to clarify what types of products are “cellular or gene therapy products” and what kinds of changes to product design constitute “different versions” for purposes of this guidance. The draft guidance specifies that these early stage trials are not expected to be powered to demonstrate a statistically significant difference in efficacy between the study arms and are not intended to provide primary evidence to support a marketing application.
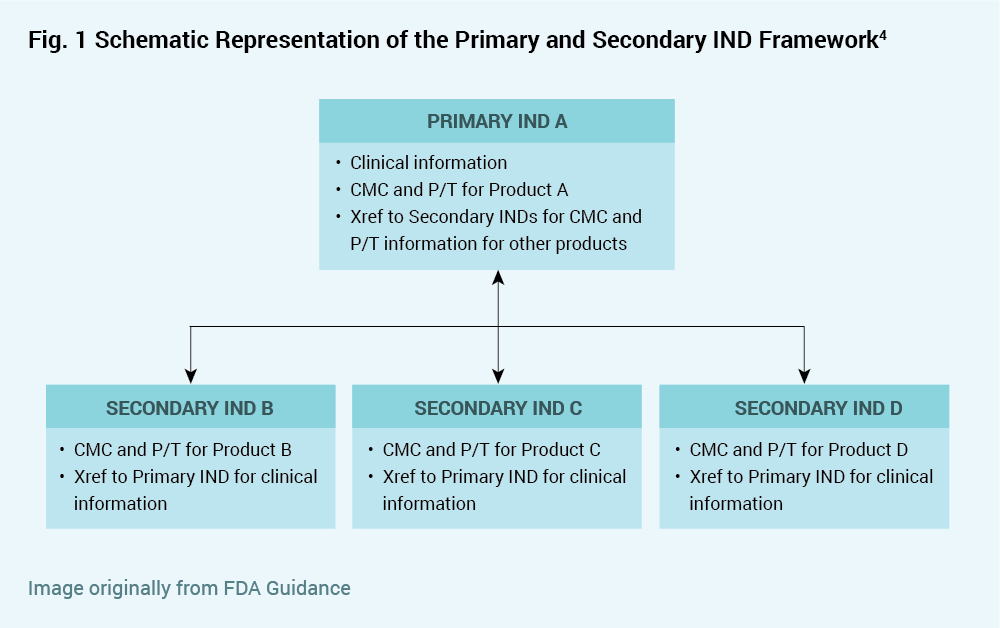
The focus of the draft guidance is to offer an orderly approach for sponsors to cross reference INDs to provide all necessary information to the FDA while minimizing submission of the same information to multiple INDs. The recommended approach is to designate a “Primary IND” that contains information about the master trial protocol, and to designate one or more “Secondary INDs” each containing specific information about the respective product version but not containing information about the umbrella trial. The draft guidance allows for various eventualities such as clinical holds, addition of new product versions, and discontinuation of clinical study for products referenced in the Primary or Secondary INDs. Safety reports regarding a specific product version must be submitted to both the Primary IND and to any Secondary IND referencing that product version (i.e. containing chemistry, manufacturing and control or pharmacology/toxicology information for that product).
Notable examples of products specifically mentioned as “cell and gene therapy products” in the Appendix include tumor infiltrating lymphocytes (TILs), chimeric antigen receptor (CAR)-T cells, dendritic cells, and gene therapy vectors. The Appendix also includes useful examples of changes to product design that constitute in new “versions” requiring a separate IND. In addition there are also examples of changes that do not result in a separate “version”—these examples are all changes at the level of the manufacturing process and the draft guidance does not suggest there is any situation where a change to the genetic sequence (of a vector, promoter/enhancer, or transgene) would not constitute a new “version.”
Overall it seems likely that this new draft guidance will be a valuable new piece in the puzzle of comprehensive gene therapy product development.
References
- [1] Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/studying-multiple-versions-cellular-or-gene-therapy-product-early-phase-clinical-trial
- [2] Master Protocols: Efficient Clinical Trial Design Strategies To Expedite Development of Oncology Drugs and Biologics, Draft Guidance for Industry, OCTOBER 2018. https://www.fda.gov/media/120721/download
- [3] Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. https://www.nejm.org/doi/full/10.1056/NEJMra1510062