
To comply with the 21st Century Cures Act,1 the Food and Drug Administration (FDA) has released two Notices of Proposed Rulemaking (NPRMs). NPRMs are the initial public notice of a proposed change to federal regulations. These two NPRMs are intended to harmonize the FDA institutional review board (IRB) and informed consent regulations with the Federal Policy for the Protection of Human Subjects (the “Common Rule”) to the extent possible.
The Common Rule was revised in 2018, and the FDA’s two recent NPRMs will harmonize the FDA regulations with most of those 2018 revisions. The Common Rule has been adopted by 20 federal agencies and applies to research that is conducted or supported by those agencies. One NPRM describes a potential requirement for multi-site research that is under FDA oversight to use a single IRB for the review of all study sites.2 The second NPRM describes changes to FDA regulations to harmonize with the Common Rule regarding informed consent requirements, among other things.3
This article will discuss how the new proposed single IRB mandate would impact institutions which currently use their local IRB for review of this research and how to prepare for compliance with the potential new regulation.
What Is the Single IRB Mandate?
If the proposed regulation is implemented, it would require any institution located in the United States (US) to rely on review and approval of a single IRB for FDA-regulated cooperative research. “Cooperative research” is defined as multi-institutional studies, multi-site studies, or multi-center studies. “Institution,” as used here, means university, community hospital, health system, or independent research site. Sponsors of FDA-regulated cooperative research will be responsible for choosing the IRB of record. Universities, hospitals, and health systems with their own IRBs will be required to cede review to the IRB of record if they wish to participate in the cooperative research.
Using a single IRB for cooperative research is not a new concept for most universities. The National Institutes of Health (NIH) led the way in announcing they would require a single IRB for multi-center research proposals in 2016, with the policy becoming effective in 2018. When the revised Common rule was finalized in 2018, it expanded this concept to research funded by the additional Common Rule Departments and Agencies. The rule became effective in January 2020.4 The FDA’s proposed rule would further expand this requirement but should not require any completely new policies or procedures for universities and other institutions already familiar with using a single IRB for federally-funded research.
Some exceptions to the proposed rule include research that requires review by more than one IRB by law (including tribal law), research involving an FDA-regulated product requiring specialized or local expertise, research on drugs exempt from the investigational new drug (IND) regulations, and research involving devices that are not subject to investigational drug exemption (IDE) regulations or are subject to abbreviated IDE requirements (non-significant risk devices). The FDA is proposing specific exceptions to the single IRB requirement rather than proposing a case-by-case assessment of each protocol due to the administrative burden such a policy would cause. The FDA requested input from stakeholders during the comment period regarding whether the exceptions proposed are sufficient or whether additional exceptions should be considered. These exceptions would be made for the entire protocol, not at a site-by-site level.
How Does External IRB Review Fit into Institutional Oversight of Research?
Research oversight at institutions often involves several offices or functions, one of which is IRB administration. The figure below provides a visual representation of how IRB review may fit into a larger constellation of research oversight at an academic institution. Even if an institution cedes IRB review to another institution or independent IRB, the institution will retain control of research oversight and will need to ensure it has procedures in place for appropriate communication between the IRB and the larger research oversight enterprise. Institutions have been working through the complexities of overseeing federally-funded research requiring review by a single IRB for several years now.
IRB Review Under the Single IRB Mandate
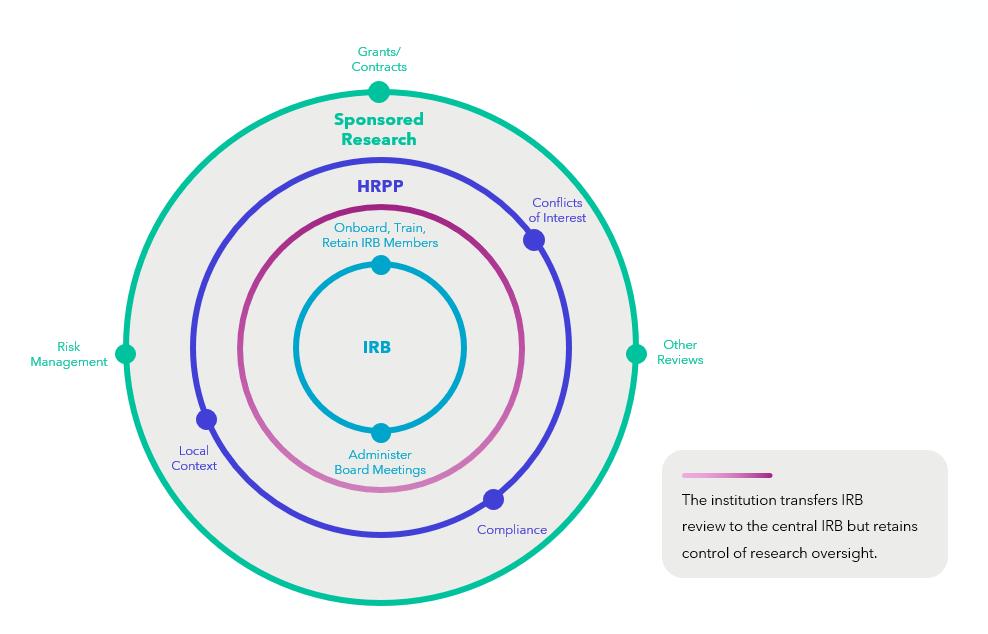
IRB Review Without the Single IRB Requirement
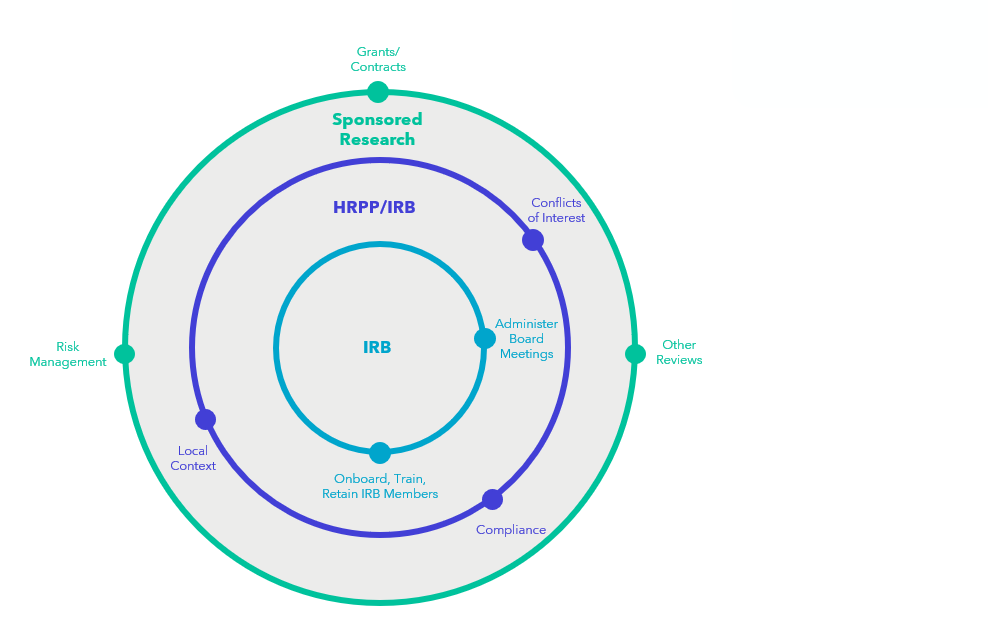
Implementation of the proposed rule might alter the balance of internally and externally reviewed research, but investigator-led, single-site research will still require support from an internal IRB unless an institution chooses to use an independent IRB. Institutional leadership may need to re-evaluate workflows and functions, including shifting staff to new purposes. For example, staff who currently support the IRB review functions might transition to a reliance team responsible for ensuring strong communication between the researchers, the internal research compliance office, and the external IRB.
How Will Investigators and Departments Be Expected to Submit to External IRBs?
Consider the requirements investigators and departments are required to meet prior to the review of your local IRB and whether the same requirements should be in place before submission to an external IRB. If you have a system in place for single IRB review for NIH-funded research, you may be able to adapt those systems to include FDA-regulated research. To make final decisions about how to manage this process, you may want to reach out to external IRBs to ask questions about their practices and policies.
Qualities to Look for in an External IRB
Accreditation: Association for the Accreditation of Human Research Protection Programs (AAHRPP) is an independent nonprofit accrediting body. AAHRPP accreditation confirms that the IRB meets industry standards for its operations and ethical reviews. You can search a list of accredited IRBs on the AAHRPP website.5
Institutional Requirements: You should ask how an external IRB collects information regarding local requirements at research sites. These requirements could include specific consent form language around conflict-of-interest management, compensation for injury, or data sharing policies. This information may be included in submission forms or instructional documents as well.
Management of Financial Conflicts of Interest: External IRB review can provide a neutral review of institutional conflicts of interest that may arise related to ownership of patent rights or equity in companies based on institutional intellectual property. You should ask about how financial conflict of interest information is obtained and reviewed by the external IRB. Ask about how any institutional determinations regarding conflict management plans should be communicated to the reviewing IRB. Once you have this information, you can begin a conversation with leadership at your institution to plan for incorporating any institutional instructions/requirements, such as consent language, into the workflow. There should also be discussion to ensure that institutional decisions, such as those related to financial conflict of interest management, are included within the panel documentation for IRB assessment.
Institutional Training: Training should be provided for Principal Investigators (PIs) and department staff who handle research administration on how to interact with the Human Research Protection Program (HRPP) office, other research reviews, and the IRB. Training on the IRB submission process for staff and PIs should also be provided so they are familiar with your new process. Training should include support for both the mechanics of how to submit a project for review and any institutional requirements you put in place.
Preparing for the Single IRB Mandate
- Re-evaluate current workflows and staff roles. Consider shifting personnel who support internal reviews to a reliance team that manages communication between researchers, the compliance office, and external review boards.
- Adapt existing systems. If your organization already handles National Institutes of Health (NIH) funded projects, modify those procedures to include Food and Drug Administration (FDA) regulated studies.
- Verify accreditation status. Look for partners accredited by the Association for the Accreditation of Human Research Protection Programs (AAHRPP) to ensure they meet ethical industry standards.
- Clarify local requirement collection. Ask potential partners how they gather specific institutional details, such as consent form language regarding injury compensation or data sharing policies.
- Understand conflict management. Request information on how an external board evaluates financial conflicts and determine how your local plans should be communicated.
- Plan new procedures with leadership. Initiate discussions with leaders to incorporate necessary instructions into the updated submission process.
- Provide comprehensive training. Educate Principal Investigators (PIs) and department personnel on new submission mechanics to guarantee they understand updated institutional requirements.
Conclusion
The federal government has been moving in the direction of requiring a single IRB to oversee multi-site research for several years. The goal of this transition is to reduce the burden on institutions and sponsors so that important research developments can reach the public more quickly without sacrificing ethical oversight.
Institutions may have concerns about the risk of ceding control over such an important aspect of research oversight. We encourage you to think about how the impending single IRB mandate fits into your current research oversight program and to reach out to ask questions early on so that you can plan carefully for this transition.
References
- 21st Century Cures Act, Section 1002, Public Law 114-255. https://www.govinfo.gov/app/details/PLAW-114publ255.
- Notice of Proposed Rulemaking: Institutional Review Boards; Cooperative Research (87 FR 58752) https://www.federalregister.gov/documents/2022/09/28/2022-21089/institutional-review-boards-cooperative-research.
- Notice of Proposed Rulemaking: Protection of Human Subjects and Institutional Review Boards (87 FR 58733). https://www.federalregister.gov/documents/2022/11/14/2022-24689/protection-of-human-subjects-and-institutional-review-boards-and-institutional-review-boards.
- Code of Federal regulations Title 45 46.114 https://www.ecfr.gov/current/title-45/subtitle-A/subchapter-A/part-46/subpart-A/section-46.114.
- AAHRPP. https://www.aahrpp.org/find-an-accredited-organization.
Related sIRB Resources

The Upcoming sIRB Mandate and the Critical Role of Site Selection: Your Questions Answered
Blog Posts
Maximizing the Power of Central IRBs Prior to the FDA’s sIRB Mandate
Videos
FDA’s Proposed Rule for Single IRB Review in Cooperative Research
Blog Posts
Revisiting the FDA’s Proposed Single IRB Mandate: Navigating Changes and Aligning for Success
VideosWho’s Supporting Your Study’s Endpoint Adjudication?
Behind every great EAC is WCG. Our experience, quality, focus, technology, and industry-leading global network put us far above the rest. Experience the WCG difference starting with a free EAC services consultation.