The Informed Consent Process
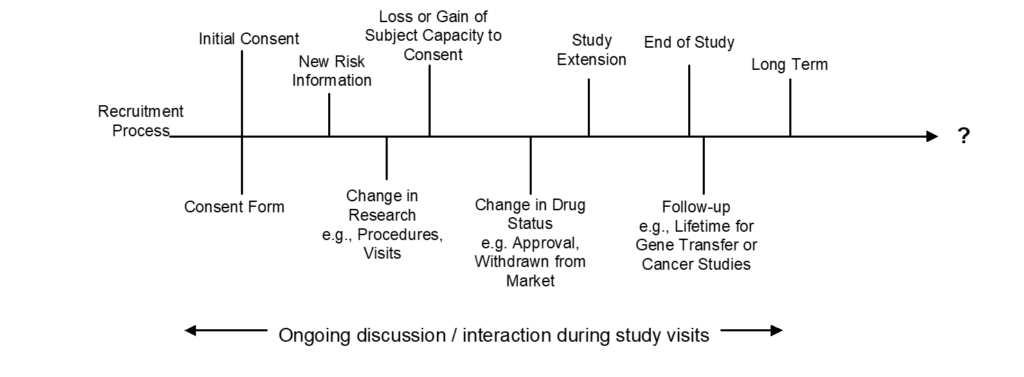
The informed consent process is central to the ethical conduct of research. It is an ongoing conversation between the human research participant and the researchers that begins before consent is given and continues until the end of the participant’s involvement in the research (see consent process diagram, below). There are various tools for the investigator to use to optimize this conversation, but the most important feature of informed consent is the investigator’s commitment to the process. More information about informed consent is available in the FDA guidance document titled “Informed Consent Guidance for IRBs, Clinical Investigators, and Sponsors.”
Goals for the Informed Consent Process
- Give the participant information about the research.
- Make sure the participant has time to consider all options.
- Answer all of the participant’s questions before the decision is made.
- Make sure that all information is understood by the participant.
- Obtain the participant’s voluntary informed consent to participate.
- Continue to inform the participant throughout the research study.
- Continue to re-affirm participant consent to participate throughout the research study.
Consent Process Diagram
Issues to Consider During the Consent Process
- Was the participant alert and, in your opinion, able to read and understand the language in the consent form?
- If the participant was unable to read the consent form, and limited or non-readers were allowed to participate, did you have an impartial witness present for the entire process? (An impartial witness is someone with adequate reading ability who is independent of the trial, who cannot be unfairly influenced by people involved in the trial, who attends the informed consent process while the consent form is being read to the participant, who reads the informed consent form and any other written information supplied to the participant, and who is willing to attest to this by signing the consent form.)
- If the participant is not fluent in English, was an approved translation of the consent form provided in the primary language of the participant? Was there also a bilingual translator present to assist with the informed consent process? Note: a translator alone is not considered adequate.
- Was the participant under any pressure (for example, family pressure, lack of medical insurance) to participate in the research? Was this discussed?
- Did the participant take time to carefully read the consent form, or read it along with you?
- Were the risks, as set forth in the consent form, carefully explained to the participant?
- Are there any other risks or concerns not stated in the consent form and were these explained to the participant?
- Was the participant asked if they had any questions about the study?
- Did the participant have any questions or concerns?
- Were the participant’s questions answered?
- Was the participant satisfied with the answer(s) they were provided?
- Did the person conducting the consent discussion check for participant understanding by asking some basic questions about the research? Did the responses reflect adequate understanding?
- Did the participant express a clear decision to proceed with the study?
- Was the consent form signed by the person who conducted the informed consent discussion?
- Was the consent form signed by a witness (if required)?
- Was the consent form signed by the principal investigator (if required)?
- If a Legally Authorized Representative is allowed to sign for the participant, were additional concerns about the participant’s understanding and assent considered and addressed?
Investigator Responsibilities Regarding Informed Consent
- Obtain consent before initiating study-specific procedures, using the current IRB approved consent form (bearing the WCG IRB approval stamp wherever feasible).
- Give the person providing informed consent as much time as they need to make a decision.
- Provide a quiet, comfortable, and private setting for the informed consent process whenever possible.
- Explain the consent process to the participant.
- Make sure the participant has time to consider all options; allow participant to take the form home before signing (whenever possible). If the person providing informed consent needs more time than is allowed by the research design, do not enroll the prospective participant.
- Evaluate whether the person providing informed consent is experiencing pressure to make a decision, and if so, do not enroll the prospective participant, even if the person providing informed consent agrees to be in the research.
- Ensure there is no threat of harm or adverse consequences to the prospective participant for a decision to not take part in the research.
- Stop the informed consent process once the person providing consent indicates that they do not want to take part in the research.
- Evaluate whether the person providing informed consent is being coerced or unduly influenced by others to take part in the research, and if so, do not enroll the prospective participant, even if the person providing informed consent agrees to be in the research.
- Communicate in the preferred language of the person providing informed consent.
- Adapt the presentation of the information to the participant’s capacities in terms of intelligence, rationality, maturity, and language.
- Invite and answer questions from the person providing informed consent.
- Evaluate whether the person providing informed consent understands the information provided and do not enroll a prospective participant who does not understand, even if that person providing informed consent agrees to be in the research.
- Ensure that no information is provided to the prospective participant or the person providing informed consent that is made to waive or appear to waive any of the prospective participant’s legal rights, or releases or appears to release the investigator, the sponsor, the institution or its agents from liability for negligence.
- Communicate to the person providing informed consent all the information in the consent document or script approved by the IRB.
- Invite and answer questions from the person providing informed consent.
- Do not enroll a prospective participant when the person obtaining informed consent is unwilling to listen to or consider the information, even if the person providing informed consent agrees to be in the research.
- Consider the participant’s reading abilities. Check to make sure the protocol does not exclude participants unable to read. If enrollment of limited or non-readers is allowed, involve an impartial witness in the informed consent process.
- Answer all questions.
- To the extent possible, make sure the participant understands enough information about the research study to give informed consent.
- To the extent possible, make sure the participant can consent free from coercion or other undue influence.
- Since the informed consent process continues throughout participation in the study, consent should be informally verified on a continuing basis.
- Significant new information must be given to the participant, and continuing consent documented in some way; for example, new risk information presented to the participant in an addendum to be signed by participants who agree to continue to participate.
Tools an Investigator Might Use to Assist the Informed Consent Process
- Consent Form – also called Informed Consent Form (ICF), Informed Consent Document (ICD) or Patient Consent Form (PCF)*
- Pamphlets or Other Reading Materials*
- Internet Information*
- Instruction Sheets*
- Audio-Visual Presentations*
- Charts or Diagrams*
- Discussions
- Consultation with Others
*These items require IRB review before use.
Ensuring Consistency of Risk Information Across Multiple Sites in a Study
When the IRB reviews and approves the template consent form in a study, it will identify the risk language that will be held consistent. This is to promote consistent disclosure of risk information across WCG sites. All site consent forms should follow the template study product risk language throughout the life of the study, including formatting when submitted. For example, if a site submits changes to the risk language and confirmation to apply the change to all sites is not received from the sponsor, additional actions may be required prior to unconditional approval.
Consent by Legally Authorized Representatives
The laws regulating who can consent for adults who lack the capacity to consent for themselves are defined at the state level and vary from state to state. Persons who can consent for adults who lack the capacity to personally provide informed consent are known as Legally Authorized Representatives (LARs). See 21 CFR 50.3(l) and 45 CFR 46.102(i). These regulations define LAR as an individual or judicial or other body authorized under applicable law to consent on behalf of a prospective participant to the participation in the procedure(s) involved in the research. We recommend you consult your state law and if necessary, obtain legal counsel to determine who represents a LAR for your research.
WCG IRB makes determinations about whether participants requiring consent by LAR are allowed in a study at the protocol level. Sites will, by default, have LAR signature lines and any associated Assent instructions/signatures on informed consent forms if this was approved for the protocol. If sites wish to enroll participants requiring consent by LAR, they will need to comply with all local laws and institutional guidelines when considering whether to enroll participants requiring consent by LAR. If a site does not want these signature lines/instructions on their consent form because they will not be enrolling them, a redlined consent form needs to be submitted for review with these lines/instructions removed.
Summary of WCG IRB’s requirements for consent form signatures when consent is required
Participant Signatures: The participant must sign and date the consent form [21 CFR 50.27(a); 45 CFR 46.117(a), ICH 4.8.8].
Signature of Person Who Conducted the Informed Consent Discussion: The person who conducts the informed consent discussion must sign and date the consent form (ICH 4.8.8).
Witness Signature: WCG IRB does not require a witness signature on the consent form, except in rare cases or as required by state or local law. However, WCG IRB will include a witness signature block at the request of the investigator or the sponsor. Because WCG IRB does not require a witness signature, WCG IRB does not have written procedures identifying who may serve as a witness. The investigator or the sponsor should have written procedures describing who may be a witness and what the witness signature signifies. If a witness signature block is included on the consent form, it must be signed for each consent form, unless the investigator’s or sponsor’s written procedures allow otherwise.
Signature of Impartial Witness: If a participant or a legally authorized representative (LAR) is unable to read because of blindness, illiteracy, or some other reason, an impartial witness should be present during the entire consent process and should sign and date the consent form in compliance with ICH E6 4.8.9. The definition of an impartial witness is provided at ICH E6 1.26. In the absence of designated signature lines, download the WCG IRB standard impartial witness form from the Download Forms page.
The definition of impartial witness is provided at ICH 1.26, which states:
ICH 1.26 “Impartial Witness – A person, who is independent of the trial, who cannot be unfairly influenced by people involved with the trial, who attends the informed consent process if the subject or the subject’s legally acceptable representative cannot read, and who reads the informed consent form and any other written information supplied to the subject.”
An impartial witness’ signature may not be used to attest to ad hoc translation of the consent into a language different than the language in which the consent form is written.
The impartial witness signature block should be left unsigned unless there is an impartial witness present for the consent process. Please see the Frequently Asked Question, “How do I obtain informed consent from someone who speaks and understands English, but cannot read English?” for further discussion of the impartial witness requirements.
Signature of Legally Authorized Representatives (LARs): In research that allows the enrollment of adult participants who are not legally competent, the consent form will include a signature block for an LAR. If an adult participant is not legally competent, a LAR must participate in the consent process, agree to the enrollment of the participant in the research, and sign the consent form. If the research allows the enrollment of adult participants who are legally competent and participants who are not legally competent, then the LAR signature block will be labeled, “when necessary.” In this case, the LAR signature block should not be signed if the participant is legally competent. The LAR signature block will only be signed when the participant is not legally competent.
Signature of Person Conducting Assent Discussion: If the participant is unable to consent due to incapacity, or because they are a minor, the person conducting the assent should also sign the consent form.
Signature of Principal Investigator: WCG IRB does not require an investigator’s signature on the consent form, only the person conducting consent; however, if a consent form is submitted with an investigator signature line, WCG IRB will not remove it, but may add a designation for the person conducting assent if not already included. If an investigator signature line is on the consent form, the investigator or an “equally qualified” sub-investigator must sign the consent form. WCG IRB does not require that the investigator’s signature date coincide with the participant’s signature date.
Consent by Participants Who Cannot Physically Sign the Consent Form
WCG IRB does not require a Legally Authorized Representative to provide consent for participants who are cognitively capable of consenting, but physically unable (for example, due to paralysis). In those cases, obtain consent from the participant with the assistance of a witness.
Consent of Cognitively Impaired Participants
You cannot enroll participants who do not have the capacity to consent unless inclusion of such adults is noted on your Certificate of Action.
If an adult participant is not capable to consent to participate in a study, WCG IRB requires that a legally authorized representative consent for the participant and in some cases also requires participants to assent to taking part in the research as well. The definition of “legally authorized representative,” as described in FDA 21 CFR § 50.3 is:
“An individual or judicial or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s participation in the procedure(s) involved in the research.”
The applicable law is the law of the state, as well as any other local law. Thus, the definition of “legally authorized representative” will be determined by state law or other local law. For questions regarding state and local law, contact a healthcare attorney admitted to the bar in that state. Changes in state and local law may occur frequently. Therefore, legal counsel should always be consulted to determine the current state of applicable law.
Obtaining Informed Consent from Someone Who Speaks and Understands English, but Cannot Read English
Sometimes potential participants speak and understand English, but cannot read due to blindness, illiteracy, or some other reason. If consent is needed for a participant in this situation, please see the section of this guide regarding possible use of a short form consenting process.
Obtaining Informed Consent from Deaf Participants
Investigators should first assess overall literacy and communicative modalities available to the participant, such as the participant’s ability to read, capability to communicate by writing, and use of American Sign Language (ASL). If the participant has literacy issues, then use of ASL is the preferred option. Investigators may utilize a translator with ASL expertise. If an impartial witness is not available, the translator can also serve as a witness; please refer to WCG’s policy on Short Forms in this guide.
Navigating Multilingual Research Intended to be Conducted Solely in a Non-English Language
WCG recognizes that there is a need to conduct studies in languages other than English, in pursuit of inclusion, and that some studies are developed never to include English speaking participants. WCG also recognizes that this presents unique challenges in the ethical review process, particularly when it comes to ensuring the accuracy and cultural relevance of translated materials. For example, if a study will be solely conducted in Spanish, ethical review is still required. Unless an Institutional Review Board (IRB) is equipped to offer ethical review services in Spanish, the documents will need to initially be submitted in English for review. Which means that materials would have to be back translated from Spanish to English for review.
Unfortunately, back translation often results in grammatical errors or changes to the original content, and additional changes made during the review process may ultimately harm the original messaging. This can result in the loss of more nuanced concepts or terminology, potentially resulting in misunderstandings or misrepresentations of the original content. In addition, direct translations may not always convey the same implications or relevance in the target language, which can compromise the cultural sensitivity and validity of the research.
Current Recommendations for Multilingual Research Review Through WCG
WCG recognizes the importance of accommodating multilingual research, and we are working to develop solutions to support our clients’ needs. While we do not currently have a mechanism for conducting ethical review of non-English materials without first reviewing the English version and thereafter approving a translation, our current recommendation for studies intended to be conducted only in a non-English language is to:
- Develop materials in the target language: Create participant-facing materials in the language that will be presented to participants.
- Translate materials into English: Translate the developed materials into English for ethical review.
- Submit with an administrative letter: Submit the translated materials with an administrative letter that requests the IRB make minimal changes to grammar and readability of the document. This letter should note that the document may not read well in English since it is intended for a non-English audience and clarify any special terms or phrases that may not have direct English equivalents.
This approach will support the development and implementation of culturally responsive and accurate participant-facing materials, acknowledging the complexities of multilingual research, while also supporting the ethical review process.
Waivers of Consent for Non-FDA Studies
If you are requesting a waiver of consent and the research is not an FDA regulated study, then criteria from 45 CFR 46.116(e) must be met:
- The research involves no more than minimal risk to the participants.
- The research could not practicably be carried out without the waiver or alteration.
- If the research involves using identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format.
- The waiver or alteration will not adversely affect the rights and welfare of the participants.
- Whenever appropriate, the participants will be provided with additional pertinent information after participation.
WCG IRB applies this standard to all requests for waiver of consent for non-FDA regulated research.
Waivers of Consent for FDA Studies
On Dec. 21, 2023, A final rule was published in the Federal Register regarding waivers of consent for FDA studies https://www.govinfo.gov/content/pkg/FR-2023-12-21/pdf/2023-27935.pdf. The final rule is effective January 22, 2024, If you are requesting a waiver of consent and the research is an FDA regulated study, then criteria from 21 CFR 50.22 must be met:
- The clinical investigation involves no more than minimal risk to the subjects;
- The clinical investigation could not practicably be carried out without the requested waiver or alteration;
- If the clinical investigation involves using identifiable private information or identifiable biospecimens, the clinical investigation could not practicably be carried out without using such information or biospecimens in an identifiable format;
- The waiver or alteration will not adversely affect the rights and welfare of the subjects; and
- Whenever appropriate, the subjects or legally authorized representatives will be provided with additional pertinent information after participation.
For individual emergency waivers of consent, prospective IRB approval is not always necessary if a patient’s life can be saved. For more information refer to 21 CFR 50.23 (a)-(c).
Waiver of Documentation of Consent
A waiver of documentation of consent is a waiver of the requirement for a signature on a consent form. The Board will need to review the information that is provided to participants to obtain consent to ensure that the required elements of consent are included in the consent discussion. If you are requesting a waiver of documentation of consent, please submit a written statement or script of this information for the Board’s review, a “participant information sheet.” To create one, use the template consent, change the title to “PARTICIPANT INFORMATION SHEET” and delete the signature blocks.
The regulations that allow the Board to approve this type of waiver:
For FDA or HHS Regulated Research (21 CFR §56.109(c)(1) and 45 CFR §46.117(c)(1)(ii)):
The research presents no more than minimal risk; and
The research involves no procedures for which written consent is normally required outside of research context.
For HHS Regulated Research (not applicable under FDA regulations) (45 CFR §46.117(c)(1)(i)):
The only record linking the participant and the research would be the consent document, and the principal risk of the research is the risk of breach of confidentiality.
Each participant will be asked whether the participant wants documentation linking the participant with the research, and the participant’s wishes will govern; and
Participants enrolling in a study under this type of waiver must be provided with the elements of consent required by the regulations and participants must consent to participate.
For HHS Regulated Research (not applicable under FDA regulations) (45 CFR §46.117(c)(1)(iii)
If the participants or legally authorized representatives are members of a distinct cultural group or community in which signing forms is not the norm, that the research presents no more than minimal risk of harm to participants and provided there is an appropriate alternative mechanism for documenting that informed consent was obtained.
California Experimental Subject’s Bill of Rights (BOR)
Submission of the California Bill of Rights (BOR) by research sites with the consent form is optional. Sites may use their own version if it contains all the information outlined in the California Health and Safety Code §24172. In addition, for sites in California, WCG automatically provides an approved BOR version along with approval documents after review. However, WCG can review and approve a BOR if submitted in the submission package and a specific request for review is made. Otherwise, these are typically just filed without review.
Additionally, according to California Health and Safety code Section 24173, a signed and dated BOR does not apply to the University of California system of institutions.