
We know complex clinical trials can accelerate drug development, offering patients timely access to transformative therapies. However, these trials have a significant impact on research sites. In this blog, we explore the data behind complex studies and look at the designs for some of those trials. In addition, we discuss the site experience, including a perspective from the University of Utah, whose team has been involved in complex clinical protocols. We will also address the role of a central institutional review board (IRB) in complex studies and offer tips for site success.
Trial Complexity by the Numbers
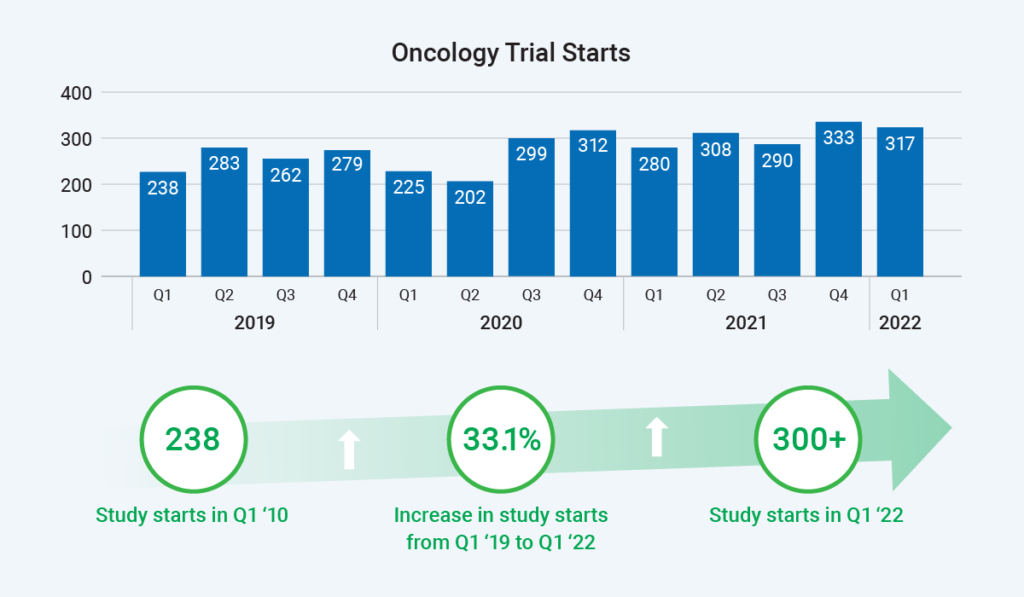
Which therapeutic area is seeing the most complex trials? Oncology research collects the greatest number of data points and involves the most trial arms. Study starts are rapidly rising in oncology, with a 33% increase from Q1 2019 to Q1 2022. Therefore, the site burden for this therapeutic area is understandably significant.
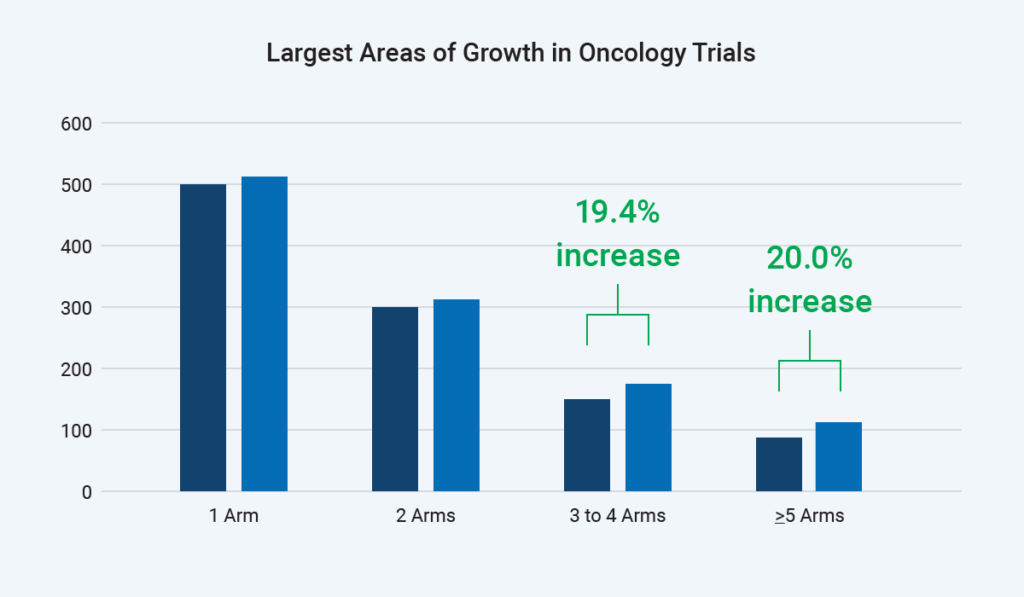
As shown, sites have seen a 20% increase in trials with three to four arms and a 20% increase in trials with four or more arms. For example, a trial may have an ovarian or breast cancer arm – or even a head and neck cancer arm – because the mechanism of action may hit several different targets across these indications. The various arms present challenges for every protocol, and we must remember that sites are managing multiple protocols.
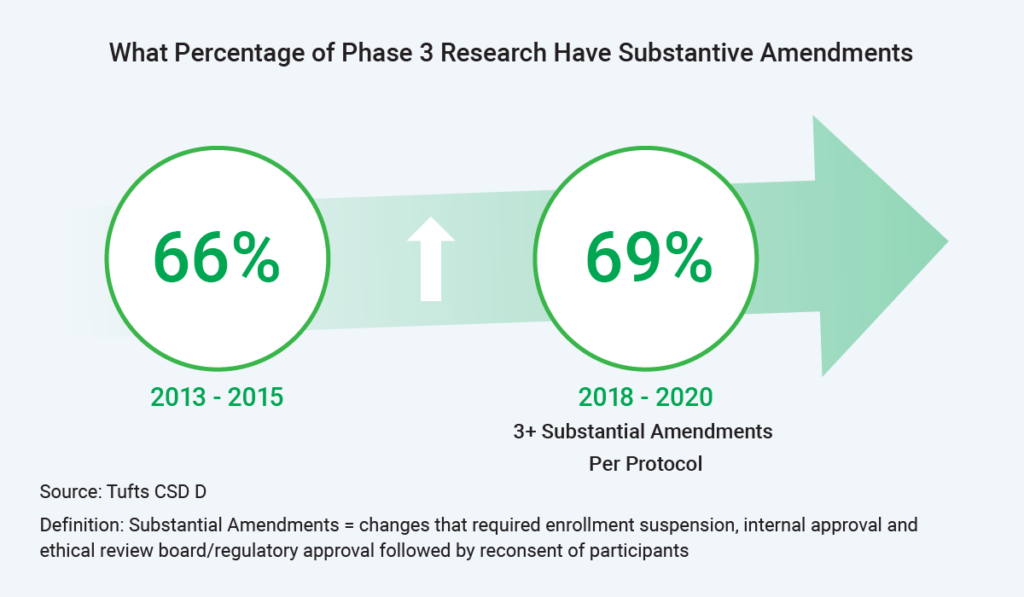
Further, sites have seen an increase in substantive amendments – from 66% to nearly 70% of Phase III trials having three or more amendments per protocol. One reason for these amendments? Enrollment is challenging, and sponsors are trying to open inclusion/exclusion criteria to increase enrollment.
Another view of trial complexity surrounds combinations of drugs. There are fewer monotherapy clinical research trials today; instead, there is a trend toward combining three or four different assets in a trial. More drugs mean more complexity, data entry requirements, and protocol length.
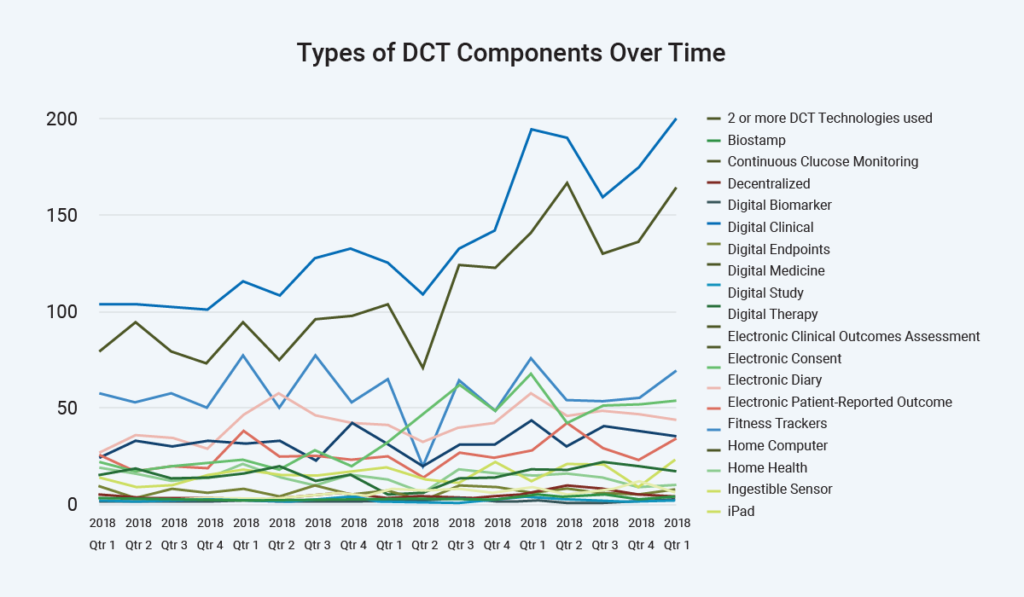
Decentralized trials are a hot topic in today’s research environment, contributing to further complexity. Sites tell us they must function as a help desk for some decentralized trials – or even hybrid trials with decentralized components. Note that decentralized definitions differ, with about 4% of research being fully virtual and the rest including components such as digital endpoints, electronic diaries, and patient-recorded outcomes.
In short, research sites today face more study starts than ever – in the most complex therapeutic area – along with more trial arms, combination therapies, and decentralized components.
Complex Protocol Designs
Let’s consider what factors make a study complex. In the traditional clinical research paradigm, we think of Phase I as looking at safety, Phase II as focusing on early efficacy indications with a small patient population, and Phase III as looking for confirmatory efficacy evidence. This view is becoming obsolete, as many studies are designed to achieve efficacy indications as early as possible – even in Phase I – for faster go/no-go decisions on whether a compound will be carried forward. Many sponsors are trying to address more questions within a single protocol to minimize the time and burden of starting a clinical study, contracting with sites, obtaining approvals, enrolling participants, analyzing the data, and so forth.
We are seeing more protocols with multiple sub-studies tacked onto the main study and more studies, including multiple amendments. Instead of stopping one study and starting the next, sponsors add a major amendment to expand cohorts, change endpoints, or change the population and keep going to minimize administrative disruption.
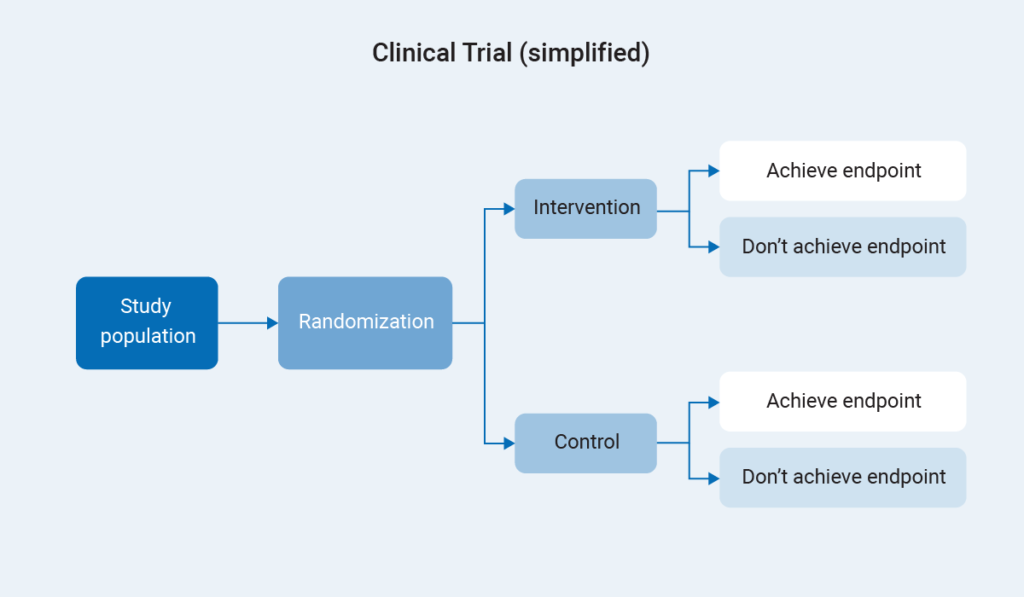
In the classic study design, a population is randomized to either an intervention or control arm. We follow both arms forward and compare the outcomes. In contrast, let’s consider the various complex studies evolving today.
Cluster randomized trials, also known as group allocation designs, do not involve study participants randomized as individuals, but at a group level- and the group may be a town, a clinic, a school or other community. For example, we could test whether hanging signs around a hospital to remind people to wash their hands affects hospital-acquired infections. Since we could not effectively remind one person and not another when they are all seeing the same signs, we might randomize by hospital or even units within a hospital for randomization at the group level rather than the individual level.
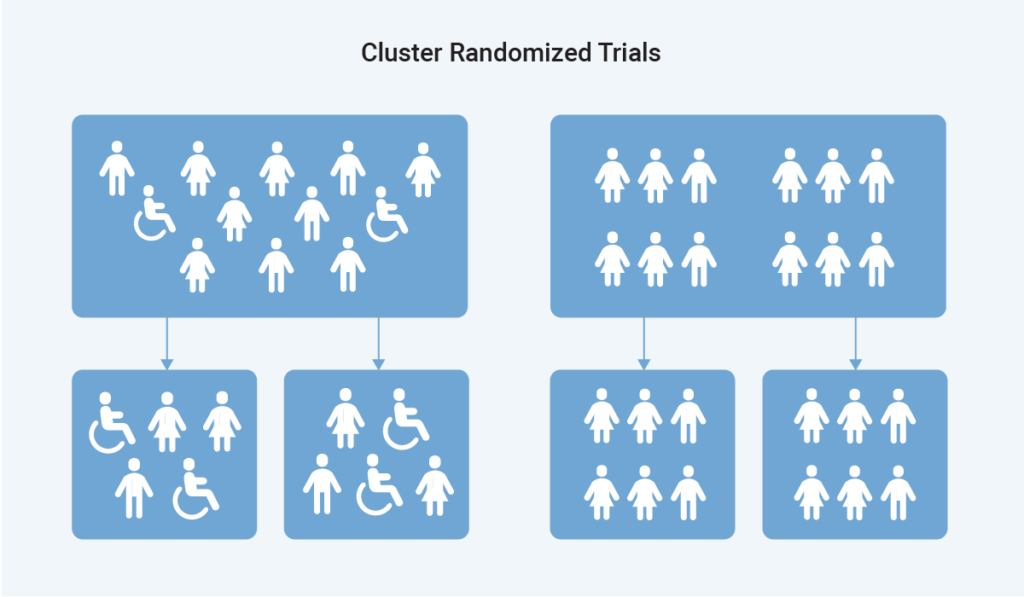
This concept applies in pragmatic studies designed to inform real decision-makers about the effectiveness of specific care interventions vs. the efficacy we measure in clinical trials. Whereas efficacy is determined in an artificial controlled clinical trial setting, effectiveness is what intervention looks like in the real world. Pragmatic trials are often cluster-randomized because they are not based on a single intervention but on how care is provided overall.
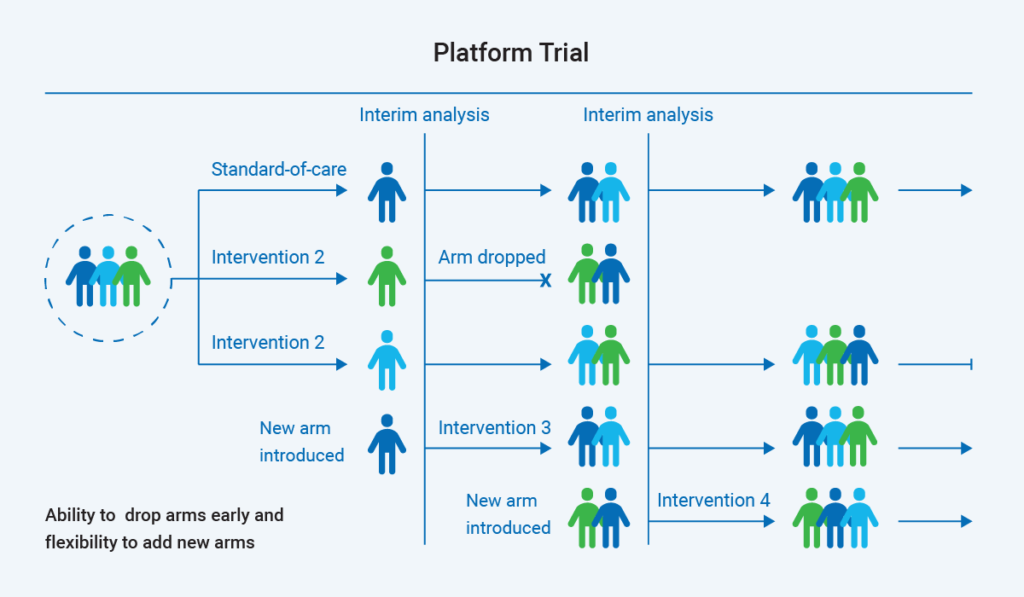
Platform trials look at multiple interventions within the context of one study. A key factor in platform studies is that they evaluate an endpoint that can be achieved fairly rapidly, because they rely on real-time or frequent interim analyses of outcome data. Arms offering potential may be continued or graduated into a separate study while other arms are dropped and more interventions added. Researchers can look at multiple interventions and answer multiple questions within a single platform and protocol without starting and stopping multiple individual studies.
“…designed for the primary purpose of informing decision-makers regarding the comparative balance of benefits, burdens and risks of a biomedical or behavioral health intervention at the individual or population level.”
Califf RM, Sugarman J. 2015. Exploring the ethical and regulatory issues in pragmatic clinical trials. Clin Trials. 12:436–441.
In addition to minimizing the operational burden of conducting multiple studies, some platforms studies are designed to adaptively randomize more people into the study arms where treatment has potential and fewer into the arms having less potential. As you can imagine, platform studies require considerable organization and coordination.
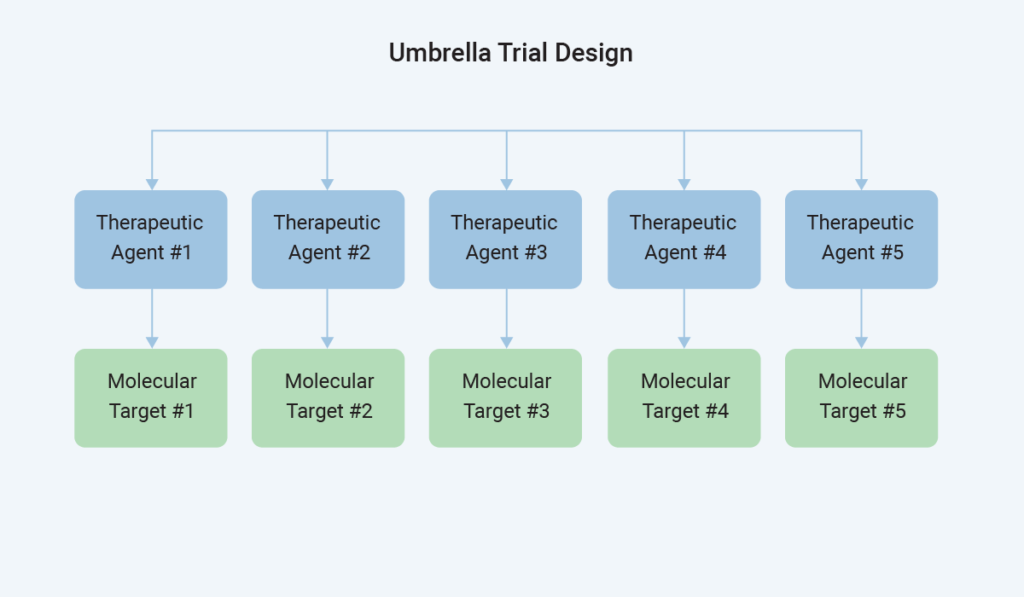
Next, consider umbrella trials – one of the master protocol designs that allow us to ask multiple questions within one study. Within a specific therapeutic indication, there are various biomarkers or molecular targets a therapeutic agent can target. For example, lung cancer study participants from different biomarker categories may go on to different interventions under the umbrella of the same clinical trial.
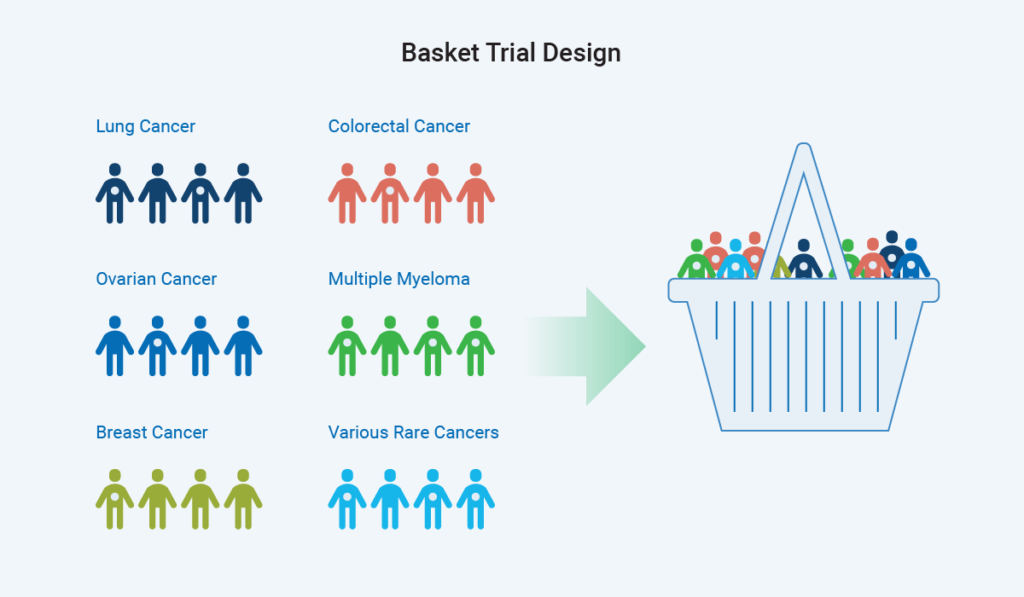
Conversely, a basket trial design looks for a biomarker or genetic mutation that is consistent across people with different indications of disease or different types of cancer. The trial then places all these people into one group, using an intervention targeted at that biomarker or pathway to measure effectiveness in those targeted populations.
Basket trials may be operationally challenging because we often perform research in therapeutic area siloes – and so must pursue new ways of thinking. In addition to being used in oncology indications, these trials may be helpful for rare genetic diseases, as genetic mutation leads to some of these diseases, and different syndromes are caused by the same mutation with different presentations. We can, therefore, put them into the same basket and address the pathway that the mutation represents.
The Site Experience
Trial complexity impacts clinical research sites dramatically – from all aspects of activation through maintenance – including enrollment. Sites may experience:
- Longer trial start-up timelines
- Higher number of protocol amendments
- Lower patient recruitment rates
- More research coordinator time
- More physician time needed
- Increased data requirements
- More technology usedAdditional training
Real-World Perspective

Jessica Moehle, CCRP, is director of the Huntsman Cancer Institute for the University of Utah. She shares the obstacles and opportunities associated with complex trials.
Benefits associated with supporting complex trials center around a higher degree of ownership for the site, including:
- Consistent and regular contact with sponsors (weekly calls)
- Ability to speak with other sites for best practice solutions
- Consistent coordination and staff feel commitment to large adaptive trials
- Knowledgeable monitors – must be proactive with quick response to sites
Challenges associated with complex trials include:
- More inconsistencies with sub-protocols and manuals of procedures, labs
- Higher volumes of protocol amendments & clarification letters
- Amendments/arms available but site not ready (committee review/approvals, pharmacy, budget work, and review needed each time)
- Some sites discontinue due to lack of response from monitor/sponsor and turnover of CRAs – adaptive trials must have consistency
Tips: Each trial is different, so sites must know how to execute appropriately. Hopefully, the site has a single contract for the entire study. Watch for additional budget work needed to address each new cohort, sub-protocol, or study arm. Training is also a critical need, with regular assessment and time scheduled to ensure that site staff are on board with procedures and ready to proceed. For example, nursing infusion pharmacies and labs may need further education on the trial design, as it will be very different from a standard clinical trial. Prerecorded training sessions will offer special value here – especially for busy physicians. Also, pay attention to vendor management and integration of technology needed to conduct complex trials.
Role of the IRB
The use of a central, independent IRB is essential for complex trials. (See the FDA recommendation in the graphic.) Why? Platform, umbrella, and basket studies are complicated, with many moving parts. Working with multiple local IRBs having multiple timelines would significantly compound the complexity.
Sites need central coordination with an IRB that can convene and review quickly, looking at unanticipated problems and new data for these adaptive studies. A central IRB will have adequate resources, expertise, and the ability to address safety data quickly.
Tips: Discuss the trial design with internal committees and request an administrative review to speed up the process. Determine up-front how the site will handle the review to pave the way for swift approvals. Also, proactive work by the sponsor to supply appropriate budgets for each sub-protocol or sub-arm will speed up the process.
Today’s evolving drug development landscape, including complex trials, presents an opportunity for accelerated discovery plus a challenge for clinical research sites.
Questions about handling complex clinical trials? Connect with WCG today.
“To facilitate IRB review of master protocols, FDA recommends the use of a central IRB. The central IRB should have adequate resources and appropriate expertise to review master protocols in a timely and thorough manner.”
“…For research conducted under a master protocol, safety data may rapidly accumulate. IRBs should consider convening additional meetings (i.e., ad hoc meetings of an existing IRB) to review the safety data that the investigator has provided to the IRB for any unanticipated problem involving risk to human subjects or others. The purpose of such meetings would be to expeditiously review the unanticipated problems and, if needed, any proposed trial modifications.”
Master Protocols: Efficient Clinical Trial Design Strategies to Expedite Development of Oncology Drugs and Biologics Guidance for Industry, FDA Guidance, March 2022
Maximize your investment: schedule a trial design and protocol planning consultation
When you have our experts review your protocol, the early investment ensures your study—and overall investment—meets its full potential. To start, simply fill out the form.